Sickle Cell Disease
Definition
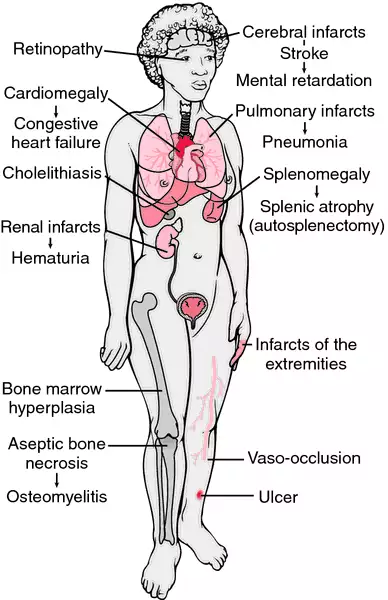
Sickle cell disease describes a group of inherited blood disorders characterized by chronic anemia, painful events, and various complications due to associated tissue and organ damage.
Because sickle cell diseases are characterized by the rapid loss of red blood cells as they enter the circulation, they are classified as hemolytic disorders, "hemolytic" referring to the destruction of the cell membrane of red blood cells resulting in the release of hemoglobin.
Description
The most common and best-known type of sickle cell disease is sickle cell anemia, which is also called meniscocytosis, sicklemia, or SS disease. All types of sickle cell disease are caused by a genetic change in hemoglobin, the oxygen-carrying protein inside the red blood cells. The red blood cells of affected individuals contain a predominance of a structural variant of the usual adult hemoglobin. This variant hemoglobin, called sickle hemoglobin, has a tendency to polymerize into rod-like structures that alter the shape of the usually flexible red blood cells. The cells take on a shape that resembles the curved blade of the sickle, an agricultural tool. Sickle cells have a shorter life span than normally shaped red blood cells. This results in chronic anemia characterized by low levels of hemoglobin and decreased numbers of red blood cells. Sickle cells are also less flexible and stickier than normal red blood cells, and can become trapped in small blood vessels preventing blood flow. This compromises the delivery of oxygen, which can result in
pain and damage to associated tissues and organs. Sickle cell disease presents with marked variability, even within families.
Carriers of the sickle cell gene are said to have sickle cell trait. Unlike sickle cell disease, sickle cell trait does not cause health problems. In fact, sickle cell trait is protective against
malaria, a disease caused by blood-borne parasites transmitted through mosquito bites. According to a widely accepted theory, the genetic mutation associated with the sickle cell trait occurred thousands of years ago. Coincidentally, this mutation increased the likelihood that carriers would survive malaria infection. Survivors then passed the mutation on to their offspring, and the trait became established throughout areas where malaria was common. As populations migrated, so did the sickle cell trait. Today, approximately one in 12 African Americans has sickle cell trait.
Worldwide, it has been estimated that one in every 250,000 babies is born annually with sickle cell disease. Sickle cell disease primarily affects people of African, Mediterranean, Middle Eastern, and Asian Indian ancestry. In the United States, sickle cell disease is most often seen in African Americans, in whom the disease occurs in one out of every 400 births. The disease has been described in individuals from several different ethnic backgrounds and is also seen with increased frequency in Latino Americans—particularly those of Caribbean, Central American, and South American ancestry. Approximately one in every 1000-1400 Latino births are affected.
Causes and symptoms
Humans normally make several types of the oxygen-carrying protein hemoglobin. An individual's stage in development determines whether he or she makes primarily embryonic, fetal, or adult hemoglobins. All types of hemoglobin are made of three components: heme, alpha (or alpha-like) globin, and beta (or beta-like) globin. Sickle hemoglobin is the result of a genetic change in the beta globin component of normal adult hemoglobin. The beta globin gene is located on chromosome 11. The sickle cell form of the beta globin gene results from the substitution of a single DNA nucleotide, or genetic building-block. The change from adenine to thymine at codon (position) 6 of the beta globin gene leads to insertion of the amino acid valine-instead of glutamic acid—at this same position in the beta globin protein. As a result of this change, sickle hemoglobin has unique properties in comparison to the usual type of adult hemoglobin.
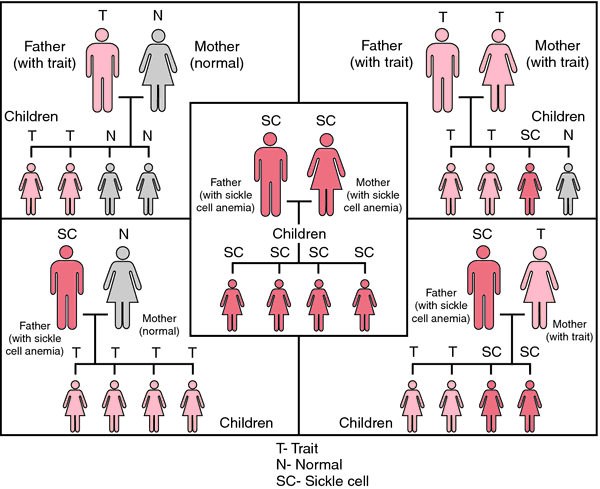
Most individuals have two normal copies of the beta globin gene, which make normal beta globin that is incorporated into adult hemoglobin. Individuals who have sickle cell trait (called sickle cell carriers) have one normal beta globin gene and one sickle cell gene. These individuals make both the usual adult hemoglobin and sickle hemoglobin in roughly equal proportions, so they do not experience any health problems as a result of having the trait. Although traces of blood in the urine and difficulty in concentrating the urine can occur, neither represents a significant health problem as a result of sickle cell trait. Of the millions of people with sickle cell trait worldwide, a small handful of individuals have experienced acute symptoms. In these very rare cases, individuals were subject to very severe physical strain.
When both members of a couple are carriers of sickle cell trait, there is a 25% chance in each
pregnancy for the baby to inherit two sickle cell genes and have sickle cell anemia, or SS disease. Correspondingly, there is a 50% chance the baby will have sickle cell trait and a 25% chance that the baby will have the usual type of hemoglobin. Other types of sickle cell disease include SC disease, SD disease, and S/beta
thalassemia. These conditions are caused by the co-inheritance of the sickle cell gene and another altered beta globin gene. For example, one parent may have sickle cell trait and the other parent may have hemoglobin C trait (another hemoglobin trait that does not cause health problems). For this couple, there would be a 25% chance of SC disease in each pregnancy.
Normal adult hemoglobin transports oxygen from the lungs to tissues throughout the body. Sickle hemoglobin can also transport oxygen. However, once the oxygen is released, sickle hemoglobin tends to polymerize (line-up) into rigid rods that alter the shape of the red blood cell. Sickling of the red blood cell can be triggered by low oxygen, such as occurs in organs with slow blood flow. It can also be triggered by cold temperatures and
dehydration.
Sickle cells have a decreased life span in comparison to normal red blood cells. Normal red blood cells survive for approximately 120 days in the bloodstream; sickle cells last only 10-12 days. As a result, the bloodstream is chronically short of red blood cells and hemoglobin, and the affected individual develops anemia.
Sickle cells can create other complications. Due to their shape, they do not fit well through small blood vessels. As an aggravating factor, the outside surfaces of sickle cells may have altered chemical properties that increase the cells' 'stickiness'. These sticky sickle cells are more likely to adhere to the inside surfaces of small blood vessels, as well as to other blood cells. As a result of the sickle cells' shape and stickiness, blockages form in small blood vessels. Such blockages prevent oxygenated blood from reaching areas where it is needed, causing pain as well as organ and tissue damage.
The severity of symptoms cannot be predicted based solely on the genetic inheritance. Some individuals with sickle cell disease develop health- or life-threatening problems in infancy, but others may have only mild symptoms throughout their lives. Individuals may experience varying degrees of health at different stages in the life cycle. For the most part, this clinical variability is unpredictable, and the reasons for the observed variability can not usually be determined. However, certain types of sickle cell disease (i.e. SC disease) tend to result in fewer and less severe symptoms on average than other types of sickle cell disease (i.e. SS disease). Some additional modifying factors are known. For example, elevated levels of fetal hemoglobin in a child or adult can decrease the quantity and severity of some symptoms and complications. Fetal hemoglobin is a normally occurring hemoglobin that usually decreases from over 90% of the total hemoglobin to under 1% during the first year of life. This change is genetically determined, although some individuals may experience elevated levels of fetal hemoglobin due to variation in the genes that control fetal hemoglobin production. Such individuals often experience a reduction in their symptoms and complications due to the ability of fetal hemoglobin to prevent the polymerization of sickle hemoglobin, which leads to sickling of the red blood cell.
There are several symptoms that warrant immediate medical attention, including the following:
- signs of infection (fever greater than >101°F or 38.3°C, coughs frequently or breathing trouble, unusual crankiness, feeding difficulties)
- signs of severe anemia (pale skin or lips, yellowing of the skin or eyes, very tired, very weak)
- signs indicating possible dehydration (vomiting, diarrhea, fewer wet diapers)
- other signs (pain or swelling in the abdomen, swollen hands or feet, screams when touched)
These can be signs of various complications that occur in sickle cell disease.
Infections and effects on the spleen
Children with sickle cell disease who are under age three are particularly prone to life-threatening bacterial infections.
Streptococcus pneumoniae is the most common offending bacteria, and invasive infection from this organism leads to
death in 15% of cases. The spleen, an organ that helps to fight bacterial infections, is particularly vulnerable to the effects of sickling. Sickle cells can impede blood flow through the spleen, causing organ damage, which usually results in loss of spleen function by late childhood. The spleen can also become enlarged due to blockages and/or increased activity of the spleen. Rapid enlargement of the spleen may be a sign of another complication called
splenic sequestration, which occurs mostly in young children and can be life-threatening. Widespread sickling in the spleen prevents adequate blood flow from the organ, removing increasing volumes of blood from the circulation and leading to accompanying signs of severe anemia.
Painful events
Painful events, also known as vaso-occlusive events, are a hallmark symptom of sickle cell disease. The frequency and duration of the pain can vary tremendously from person to person and over an individual's life cycle. Painful events are the most common cause of hospitalizations in sickle cell disease. However, only a small portion of individuals with sickle cell disease experience frequent and severe painful events. Most painful events can be managed at home. Pain results when small blood vessel blockages prevent oxygen from reaching tissues. Pain can affect any area of the body, although the extremities, chest, abdomen, and bones are frequently affected sites. There is some evidence that cold temperatures or infection can trigger a painful event, but most events occur for unknown reasons. The hand-foot syndrome, or dactylitis, is a particular type of painful event. Most common in toddlers, dactylitis results in pain and swelling in the hands and feet, sometimes accompanied by a fever.
Anemia
Sickle cells have a high turnover rate leading to a deficit of red blood cells in the bloodstream. Common symptoms of anemia include
fatigue, paleness, and a
shortness of breath. A particularly severe form of anemia—aplastic anemia—occurs following infection with parvovirus. Parvovirus causes extensive destruction of the bone marrow, bringing production of new red blood cells to a halt. Bone marrow production resumes after seven to 10 days; however, given the short lives of sickle cells, even a brief shut-down in red blood cell production can cause a rapid decline in hemoglobin concentrations.
Delayed growth
The energy demands of the bone marrow for red blood cell production compete with the demands of a growing body. Children with sickle cell anemia may have delayed growth and reach
puberty at a later age than normal. By early adulthood, they catch up on growth and attain normal height; however, weight typically remains below average.
Stroke
Children with sickle cell disease have a significantly elevated risk of having a
stroke, which can be one of the most concerning complications of sickle cell disease. Approximately 11% of individuals with sickle cell disease will have a recognizable stroke by the age of 20.
Magnetic resonance imaging studies have found that 17% of children with sickle cell anemia have evidence of a previous stroke or clinically 'silent' stroke-like events called
transient ischemic events. Stroke in sickle cell disease is usually caused by a blockage of a blood vessel, but about one fourth of the time may be caused by a hemorrhage (or rupture) of a blood vessel.
Strokes result in compromised delivery of oxygen to an area of the brain. The consequences of stroke can range from life-threatening, to severe physical or cognitive impairments, to apparent or subtle learning disabilities, to undetectable effects. Common stroke symptoms include weakness or numbness that affects one side of the body, sudden behavioral changes, loss of vision, confusion, loss of speech or the ability to understand spoken words,
dizziness, headache, seizures, vomiting, or even
coma.
Approximately two-thirds of the children who have a stroke will have at least one more. Transfusions have been shown to decrease the incidence of a second stroke. A recent study showed that children at highest risk to experience a first stroke were 10 times more likely to stroke if untreated when compared to high-risk children treated with chronic blood
transfusion therapy. High-risk children were identified using transcranial doppler ultrasound technology to detect individuals with increased blood flow speeds due to constricted intracranial blood vessels.
As of 2003, researchers are investigating various techniques for helping children with memory loss related to strokes caused by sickle cell disease.
Acute chest syndrome
Acute chest syndrome (ACS) is a leading cause of death for individuals with sickle cell disease, and recurrent attacks can lead to permanent lung damage. Therefore rapid diagnosis and treatment is of great importance. ACS can occur at any age and is similar but distinct from
pneumonia. Affected persons may experience fever,
cough, chest pain, and shortness of breath. ACS seems to have multiple causes including infection, sickling in the small blood vessels of the lungs, fat embolisms to the lungs, or a combination of factors.
Priapism
Males with sickle cell anemia may experience
priapism, a condition characterized by a persistent and painful erection of the penis. Due to blood vessel blockage by sickle cells, blood is trapped in the tissue of the penis. Priapism may be short in duration or it may be prolonged. Priapism can be triggered by low oxygen (hypoxemia), alcohol consumption, or sexual intercourse. Since priapism can be extremely painful and result in damage to this tissue causing
impotence, rapid treatment is essential.
Kidney disease
The environment in the kidney is particularly prone to damage from sickle cells. Signs of kidney damage can include blood in the urine, incontinence, and enlarged kidneys. Adults with sickle cell disease often experience insufficient functioning of the kidneys, which can progress to kidney failure in a small percentage of adults with sickle cell disease.
Jaundice and gallstones
Jaundice is indicated by a yellow tone in the skin and eyes, and alone it is not a health concern. Jaundice may occur if bilirubin levels increase, which can occur with high levels of red blood cell destruction. Bilirubin is the final product of hemoglobin degradation, and is typically removed from the bloodstream by the liver. Therefore, jaundice can also be a sign of a poorly functioning liver, which may also be evidenced by an enlarged liver. Increased bilirubin also leads to increased chance for
gallstones in children with sickle cell disease. Treatment, which may include removal of the gall bladder, may be selected if the gallstones start causing symptoms.
Retinopathy
The blood vessels that supply oxygen to the retina—the tissue at the back of the eye—may be blocked by sickle cells, leading to a condition called retinopathy. This is one of the only complications that is actually more common in SC disease as compared to SS disease. Retinopathy can be identified through regular ophthalmology evaluations and effectively treated in order to avoid damage to vision.
Joint problems
Avascular necrosis of the hip and shoulder joints, in which bone damage occurs due to compromised blood flow due to sickling, can occur later in childhood. This complication can affect an individual's physical abilities and result in substantial pain.
Diagnosis
Inheritance of sickle cell disease or trait cannot be prevented, but it may be predicted. Screening is recommended for individuals in high-risk populations. In the United States, African Americans and Latino Americans have the highest risk of having the disease or trait. Sickle cell is also common among individuals of Mediterranean, Middle Eastern, and Eastern Indian descent.
A complete
blood count (CBC) will describe several aspects of an individual's blood cells. A person with sickle cell disease will have a lower than normal hemoglobin level, together with other characteristic red blood cell abnormalities. A
hemoglobin electrophoresis is a test that can help identify the types and quantities of hemoglobin made by an individual. This test uses an electric field applied across a slab of gellike material. Hemoglobins migrate through this gel at various rates and to specific locations, depending on their size, shape, and electrical charge. Although sickle hemoglobin (Hb S) and regular adult hemoglobin (called Hb A) differ by only one amino acid, they can be clearly separated using
hemoglobin electrophoresis.
Isoelectric focusing and
high-performance liquid chromatography (HPLC) use similar principles to separate hemoglobins and can be used instead of or in various combinations with hemoglobin electrophoresis to determine the types of hemoglobin present.
Another test called the sickledex can help confirm the presence of sickle hemoglobin, although this test cannot provide accurate or reliable diagnosis when used alone. When Hb S is present, but there is an absence or only a trace of Hb A, sickle cell anemia is a likely diagnosis. Additional beta globin DNA testing, which looks directly at the beta globin gene, can be performed to help confirm the diagnosis and establish the exact genetic type of sickle cell disease. CBC and hemoglobin electrophoresis are also typically used to diagnose sickle cell trait and various other types of beta globin traits.
Diagnosis of sickle cell disease can occur under various circumstances. If an individual has symptoms that are suggestive of this diagnosis, the above-described screening tests can be performed followed by DNA testing, if indicated. Screening at birth using HPLC or a related technique offers the opportunity for early intervention. More than 40 states include sickle cell screening as part of the usual battery of blood tests done for newborns. This allows for early identification and treatment. Hemoglobin trait screening is recommended for any individual of a high-risk ethnic background who may be considering having children. When both members of a couple are found to have sickle cell trait, or other related hemoglobin traits, they can receive
genetic counseling regarding the risk of sickle cell disease in their future children and various testing options.
Sickle cell disease can be identified before birth through the use of prenatal diagnosis.
Chorionic villus sampling (CVS) can be offered as early as 10 weeks of pregnancy and involves removing a sample of the placenta made by the baby and testing the cells. CVS carries a risk of causing a
miscarriage that is between one-half to one percent.
Amniocentesis is generally offered between 15 and 22 weeks of pregnancy, but can sometimes be offered earlier. Two to three tablespoons of the fluid surrounding the baby is removed. This fluid contains fetal cells that can be tested. This test carries a risk of causing a miscarriage, which is not greater than one percent. Pregnant woman and couples may choose prenatal testing in order to prepare for the birth of a baby that may have sickle cell disease. Alternately, knowing the diagnosis during pregnancy allows for the option of pregnancy termination.
Preimplantation genetic diagnosis (PGD) is a relatively new technique that involves in-vitro fertilization followed by
genetic testing of one cell from each developing embryo. Only the embryos unaffected by sickle cell disease are transferred back into the uterus. PGD is currently available on a research basis only, and is relatively expensive.
Treatment
There are several practices intended to prevent some of the symptoms and complications of sickle cell disease. These include preventative
antibiotics, good hydration, immunizations, and access to comprehensive care. Maintaining good health through adequate
nutrition, avoiding stresses and infection, and getting proper rest is also important. Following these guidelines is intended to improve the health of individuals with sickle cell disease.
Penicillin
Infants are typically started on a course of penicillin that extends from infancy to age six. Use of this antibiotic is meant to ward off potentially fatal infections. Infections at any age are treated aggressively with antibiotics. Vaccines for common infections, such as pneumococcal pneumonia, are also recommended.
Pain management
Pain is one of the primary symptoms of sickle cell anemia, and controlling it is an important concern. The methods necessary for pain control are based on individual factors. Some people can gain adequate pain control through over-the-counter oral painkillers (
analgesics). Other individuals, or painful events, may require stronger methods that can include administration of
narcotics. Alternative therapies may be useful in avoiding or controlling pain, including relaxation, hydration, avoiding extremes of temperature, and the application of local warmth.
Blood transfusions
Blood transfusions are not usually given on a regular basis but are used to treat individuals with frequent and severe painful events, severe anemia, and other emergencies. In some cases blood transfusions are given as a preventative measure, for example to treat spleen enlargement or prevent a second stroke (or a first stroke in an individual shown to be at high risk).
Regular blood transfusions have the potential to decrease formation of hemoglobin S, and reduce associated symptoms. However, there are limitations and risks associated with regular blood transfusions, including the risk of blood-borne infection and sensitization to proteins in the transfused blood that can make future transfusions very difficult. Most importantly, chronic blood transfusions can lead to iron overload. The body tends to store excess iron, such as that received through transfusions, in various organs. Over time, this iron storage can cause damage to various tissues and organs, such as the heart and endocrine organs.
Some of this damage can be prevented by the administration of a medication called desferoxamine that helps the body to eliminate excess iron through the urine. Alternately, some individuals receive a new, non-standard treatment called erythrocytapheresis. This involves the automated removal of sickle cells and is used in conjunction with a reduced number of regular transfusions. This treatment helps to reduce iron overload.
Hydroxyurea
Emphasis is being placed on developing drugs that treat sickle cell anemia directly. The most promising of these drugs in the late 1990s is hydroxyurea, a drug that was originally designed for anticancer treatment. Hydroxyurea has been shown to reduce the frequency of painful crises and acute chest syndrome in adults, and to lessen the need for blood transfusions. Hydroxyurea seems to work by inducing a higher production of fetal hemoglobin. The major side effects of the drug include decreased production of platelets, red blood cells, and certain white blood cells. The effects of long-term hydroxyurea treatment are unknown; however, a nine-year follow-up study of 299 adults with frequent painful crises reported in 2003 that taking hydroxyurea was associated with a 40% reduction in mortality.
Bone marrow transplantation
Bone marrow transplantation has been shown to cure sickle cell anemia in some cases. This treatment is reserved primarily for severely affected children with a healthy donor whose marrow proteins match those of the recipient, namely a brother or sister who has inherited the same tissue type. Indications for a bone marrow transplant are stroke, recurrent acute chest syndrome, and chronic unrelieved pain.
Bone marrow transplantations tend to be the most successful in children; adults have a higher rate of transplant rejection and other complications. There is approximately a 10% fatality rate associated with bone marrow transplants done for sickle cell disease. Survivors face potential long-term complications, such as chronic graft-versus-host disease (an immunemediated attack by the donor marrow against the recipient's tissues),
infertility, and development of some forms of
cancer. A relatively recent advance in transplantation involves the use of donor stem cells obtained from
cord blood, the blood from the placenta that is otherwise discarded following the birth of a new baby. Cord blood cells, as opposed to fully mature bone marrow cells, appear to be less likely to result in graft-versus-host disease in the recipient. This increases the safety and efficacy of the transplant procedure.
Surgery
Certain surgical interventions are utilized in the treatment of specific sickle cell-related complications. Removal of a dysfunctioning gallbladder or spleen can often lead to improvements in health. Investigations are currently underway to establish the efficacy of hip coring surgery, in which a portion of affected bone is removed to treat avascular necrosis of the hip. The hope is that this may provide an effective treatment to alleviate some pain and restore function in the affected hip.
Gene research
Replacing the gene that produces the defective hemoglobin in sickle cell disease patients with one that makes normal hemoglobin may be a possible treatment due to recent research. According to a 1998 report in Science, researchers studied the blood cells from people who carry the sickle cell gene. By using an enzyme called a ribosome, the study was able to alter sickle cells into normal cells. The ribosome cut out the mutated instructions in the cells' genetic pattern and replaced them with the correct instructions. Researchers hope that this will allow the cells to make normal hemoglobin—leading to the ultimate treatment for those with sickle cell disease.
In late 2001, genetic scientists reported that they had designed a gene that might lead to a future treatment of sickle cell anemia. Although the gene had not been tested in humans, early results showed that the injected gene protected cells from sickling. As of 2003,
Key terms
Amino acid — Organic compounds that form the building blocks of protein. There are 20 types of amino acids (eight are "essential amino acids" which the body cannot make and must therefore be obtained from food).
Anemia — A blood condition in which the level of hemoglobin or the number of red blood cells falls below normal values. Common symptoms include paleness, fatigue, and shortness of breath.
Bilirubin — A yellow pigment that is the end result of hemoglobin breakdown. This pigment is metabolized in the liver and excreted from the body through the bile. Bloodstream levels are normally low; however, extensive red cell destruction leads to excessive bilirubin formation and jaundice.
Bone marrow — A spongy tissue located in the hollow centers of certain bones, such as the skull and hip bones. Bone marrow is the site of blood cell generation.
Bone marrow transplantation — A medical procedure used to treat some diseases that arise from defective blood cell formation in the bone marrow. Healthy bone marrow is extracted from a donor to replace the marrow in an ailing individual. Proteins on the surface of bone marrow cells must be identical or very closely matched between a donor and the recipient.
Globin — One of the component protein molecules found in hemoglobin. Normal adult hemoglobin has a pair each of alpha-globin and beta-globin molecules.
Heme — The iron-containing molecule in hemoglobin that serves as the site for oxygen binding.
Hemoglobin — Protein-iron compound in the blood that carries oxygen to the cells and carries carbon dioxide away from the cells.
Hemoglobin A — Normal adult hemoglobin that contains a heme molecule, two alpha-globin molecules, and two beta-globin molecules.
Hemoglobin S — Hemoglobin produced in association with the sickle cell trait; the beta-globin molecules of hemoglobin S are defective.
Hemolytic — Referring to the destruction of the cell membranes of red blood cells, resulting in the release of hemoglobin from the damaged cell.
Hydroxyurea — A drug that has been shown to induce production of fetal hemoglobin. Fetal hemoglobin has a pair of gamma-globin molecules in place of the typical beta-globins of adult hemoglobin. Higher-than-normal levels of fetal hemoglobin can prevent sickling from occurring.
Impotence — The inability to have a penile erection, which can be due to tissue damage resulting from sickling within the penis (priapism).
Iron overload — A side effect of frequent blood transfusions in which the body accumulates abnormally high levels of iron. Iron deposits can form in organs, particularly the heart, and cause life-threatening damage.
Jaundice — Yellowing of the skin or eyes due to excess of bilirubin in the blood.
Magnetic resonance imaging (MRI) — A technique that employs magnetic fields and radio waves to create detailed images of internal body structures and organs, including the brain.
Mutation — A permanent change in the genetic material that may alter a trait or characteristic of an individual, or manifest as disease, and can be transmitted to offspring.
Narcotics — Strong, prescription medication that can be effective in treating pain, but have the potential to be habit-forming if their use is not supervised correctly.
Nucleic acid — The cellular molecules DNA and RNA that act as coded instructions for the production of proteins and are copied for transmission of inherited traits.
Placenta — The organ responsible for oxygen and nutrition exchange between a pregnant mother and her developing baby.
Red blood cell — Hemoglobin-containing blood cells that transport oxygen from the lungs to tissues. In the tissues, the red blood cells exchange their oxygen for carbon dioxide, which is brought back to the lungs to be exhaled.
Screening — Process through which carriers of a trait may be identified within a population.
Sickle cell — A red blood cell that has assumed an elongated shape due to the presence of hemoglobin S.
experiments in
gene therapy for sickle cell disease have been carried out in mice, using lentiviral vectors to transfer the corrective gene into the mouse's stem cells. This technique, however, has not yet been attempted in human subjects as of late 2003.
Psychosocial support
As in any lifelong, chronic disease, comprehensive care is important. Assistance with the emotional, social, family-planning, economic, vocational, and other consequences of sickle cell disease can enable affected individuals to better access and benefit from their medical care.
Prognosis
Sickle cell disease is characteristically variable between and within affected individuals. Predicting the course of the disorder based solely on genes is not possible. Several factors aside from genetic inheritance determine the prognosis for affected individuals, including the frequency, severity, and nature of specific complications in any given individual. The availability and access of comprehensive medical care also plays an important role in preventing and treating serious, acute complications, which cause the majority of sickle cell-related deaths. For those individuals who do not experience such acute events, life expectancy is probably substantially greater than the average for all people with sickle cell disease. The impact of recent medical advances supports the hypothesis that current life expectancies may be significantly greater than those estimated in the early 1990s. At that time, individuals with SS disease lived to the early- to mid-40s, and those with SC disease lived into the upper 50s on average. As of 2003, the life expectancy of persons with SS is over 50. With early detection and comprehensive medical care, most people with sickle cell disease are in fairly good health most of the time. Most individuals can be expected to live well into adulthood, enjoying an improved quality of life including the ability to choose a variety of education, career, and family-planning options for themselves.
Resources
Books
Beers, Mark H., MD, and Robert Berkow, MD., editors. "Anemias Caused by Excessive Hemolysis: Sickle Cell Diseases." Section 11, Chapter 127 In The Merck Manual of Diagnosis and Therapy. Whitehouse Station, NJ: Merck Research Laboratories, 2004.
Beers, Mark H., MD, and Robert Berkow, MD., editors. "Pregnancy Complicated by Disease: Hemoglobinopathies." Section 18, Chapter 251 In The Merck Manual of Diagnosis and Therapy. Whitehouse Station, NJ: Merck Research Laboratories, 2004.
Periodicals
Davies, S. C., and A. Gilmore. "The Role of Hydroxyurea in the Management of Sickle Cell Disease." Blood Reviews 17 (June 2003): 99-109.
Egbert Maikler, Virginia, et al. "Children's and Adolescents' Use of Diaries for Sickle Cell Pain." Journal of the Society of Pediatric Nurses 6, no. 4 (October—December 2001): 161-169.
Harris, Leslie. "Living Well with Sickle Cell." Essence September 1999: 58.
Nienhuis,A. W., H. Hanawa, N. Sawai, et al. "Development of Gene Therapy for Hemoglobin Disorders." Annals of the New York Academy of Science 996 (May 2003): 101-111.
Seppa, N. "Gene Therapy for Sickle-cell Disease?" Science News 160, no. 24 (December 15, 2001): 372.
Steinberg, M. H., F. Barton, O. Castro, et al. "Effect of Hydroxyurea on Mortality and Morbidity in Adult Sickle Cell Anemia: Risks and Benefits up to 9 Years of Treatment." Journal of the American Medical Association 289 (April 2, 2003): 1645-1651.
Winrow, N., and E. R. Melhem. "Sickle Cell Disease and Stroke in a Pediatric Population. Evidence-Based Diagnostic Evaluation." Neuroimaging Clinics of North America 13 (May 2003): 185-196.
Yerys, B. E., D. A. White, C. F. Salorio, et al. "Memory Strategy Training in Children with Cerebral Infarcts Related to Sickle Cell Disease." Journal of Pediatric Hematology and Oncology 25 (June 2003): 495-498.
Organizations
Mayo Foundation for Medical Education and Research. http://www.mayohealth.org.
Sickle Cell Disease Association of America. 200 Corporate Point, Suite 495, Culver City, CA 90230-7633. (310) 216-6363. (800) 421-8453. 〈http://sicklecelldisease.org/〉.
Sickle Cell Disease Program, Division of Blood Diseases and Resources. National Heart, Lung, and Blood Institute. II Rockledge Centre, 6701 Rockledge Dr. MSC 7950, Bethesda, MD 20892-7950. (301) 435-0055.
Gale Encyclopedia of Medicine. Copyright 2008 The Gale Group, Inc. All rights reserved.